2.2. Manifold learning¶
Manifold learning is an approach to non-linear dimensionality reduction. Algorithms for this task are based on the idea that the dimensionality of many data sets is only artificially high.
2.2.1. Introduction¶
High-dimensional datasets can be very difficult to visualize. While data in two or three dimensions can be plotted to show the inherent structure of the data, equivalent high-dimensional plots are much less intuitive. To aid visualization of the structure of a dataset, the dimension must be reduced in some way.
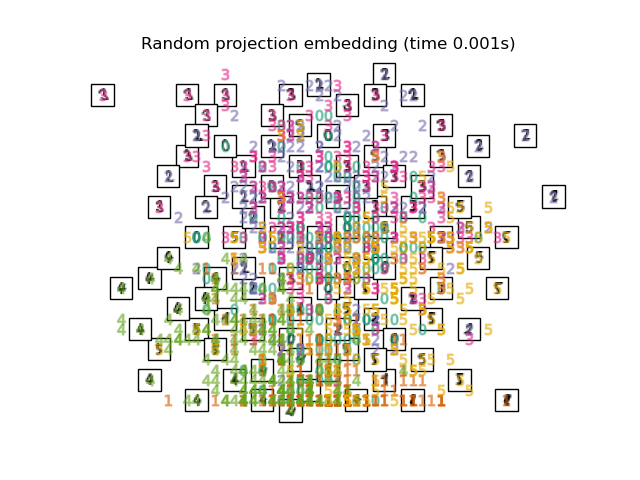
The simplest way to accomplish this dimensionality reduction is by taking a random projection of the data. Though this allows some degree of visualization of the data structure, the randomness of the choice leaves much to be desired. In a random projection, it is likely that the more interesting structure within the data will be lost.

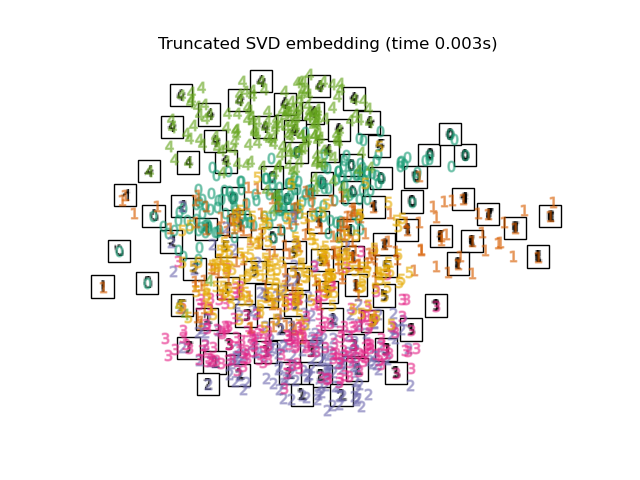
To address this concern, a number of supervised and unsupervised linear dimensionality reduction frameworks have been designed, such as Principal Component Analysis (PCA), Independent Component Analysis, Linear Discriminant Analysis, and others. These algorithms define specific rubrics to choose an “interesting” linear projection of the data. These methods can be powerful, but often miss important non-linear structure in the data.
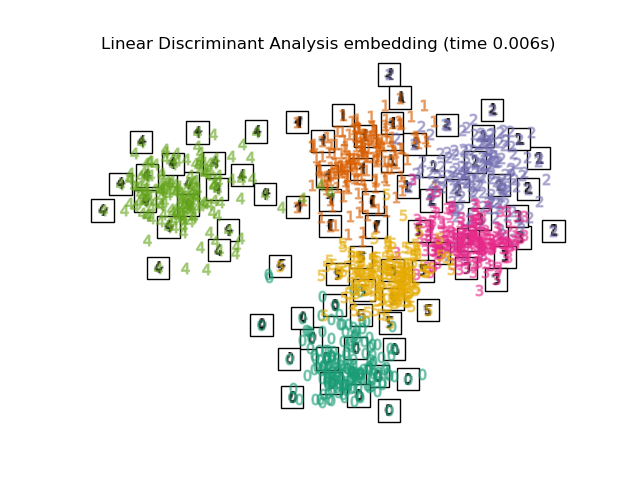
Manifold Learning can be thought of as an attempt to generalize linear frameworks like PCA to be sensitive to non-linear structure in data. Though supervised variants exist, the typical manifold learning problem is unsupervised: it learns the high-dimensional structure of the data from the data itself, without the use of predetermined classifications.
The manifold learning implementations available in scikit-learn are summarized below
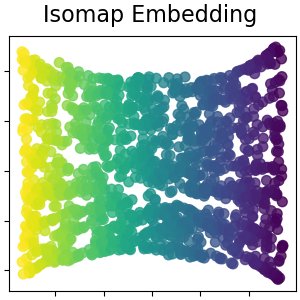
2.2.2. Isomap¶
One of the earliest approaches to manifold learning is the Isomap
algorithm, short for Isometric Mapping. Isomap can be viewed as an
extension of Multi-dimensional Scaling (MDS) or Kernel PCA.
Isomap seeks a lower-dimensional embedding which maintains geodesic
distances between all points. Isomap can be performed with the object
Isomap.
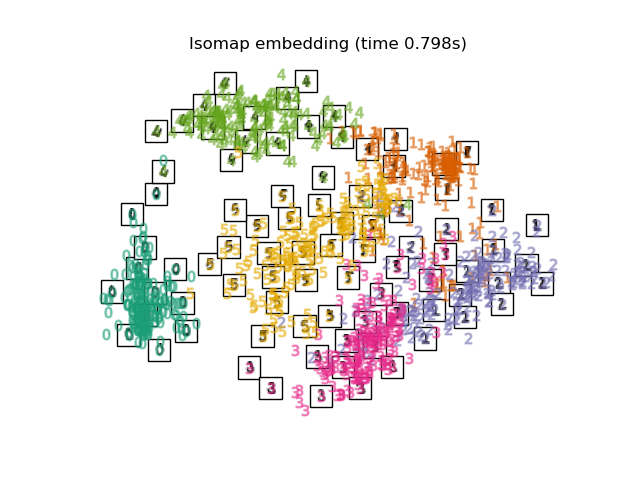
The Isomap algorithm comprises three stages: Nearest neighbor search. Isomap uses
Shortest-path graph search. The most efficient known algorithms
for this are Dijkstra’s Algorithm, which is approximately
\(O[N^2(k + \log(N))]\), or the Floyd-Warshall algorithm, which
is \(O[N^3]\). The algorithm can be selected by the user with
the Partial eigenvalue decomposition. The embedding is encoded in the
eigenvectors corresponding to the \(d\) largest eigenvalues of the
\(N \times N\) isomap kernel. For a dense solver, the cost is
approximately \(O[d N^2]\). This cost can often be improved using
the The overall complexity of Isomap is
\(O[D \log(k) N \log(N)] + O[N^2(k + \log(N))] + O[d N^2]\). \(N\) : number of training data points \(D\) : input dimension \(k\) : number of nearest neighbors \(d\) : output dimension
Complexity
Click for more details
BallTree for efficient neighbor search.
The cost is approximately \(O[D \log(k) N \log(N)]\), for \(k\)
nearest neighbors of \(N\) points in \(D\) dimensions.path_method keyword of Isomap. If unspecified, the code
attempts to choose the best algorithm for the input data.ARPACK solver. The eigensolver can be specified by the user
with the eigen_solver keyword of Isomap. If unspecified, the
code attempts to choose the best algorithm for the input data.
2.2.3. Locally Linear Embedding¶
Locally linear embedding (LLE) seeks a lower-dimensional projection of the data which preserves distances within local neighborhoods. It can be thought of as a series of local Principal Component Analyses which are globally compared to find the best non-linear embedding.
Locally linear embedding can be performed with function
locally_linear_embedding or its object-oriented counterpart
LocallyLinearEmbedding.
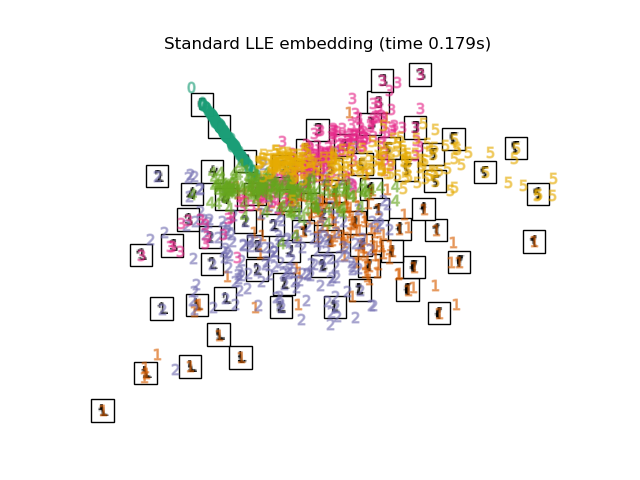
The standard LLE algorithm comprises three stages: Nearest Neighbors Search. See discussion under Isomap above. Weight Matrix Construction. \(O[D N k^3]\).
The construction of the LLE weight matrix involves the solution of a
\(k \times k\) linear equation for each of the \(N\) local
neighborhoods Partial Eigenvalue Decomposition. See discussion under Isomap above. The overall complexity of standard LLE is
\(O[D \log(k) N \log(N)] + O[D N k^3] + O[d N^2]\). \(N\) : number of training data points \(D\) : input dimension \(k\) : number of nearest neighbors \(d\) : output dimension
Complexity
Click for more details
2.2.4. Modified Locally Linear Embedding¶
One well-known issue with LLE is the regularization problem. When the number of neighbors is greater than the number of input dimensions, the matrix defining each local neighborhood is rank-deficient. To address this, standard LLE applies an arbitrary regularization parameter \(r\), which is chosen relative to the trace of the local weight matrix. Though it can be shown formally that as \(r \to 0\), the solution converges to the desired embedding, there is no guarantee that the optimal solution will be found for \(r > 0\). This problem manifests itself in embeddings which distort the underlying geometry of the manifold.
One method to address the regularization problem is to use multiple weight
vectors in each neighborhood. This is the essence of modified locally
linear embedding (MLLE). MLLE can be performed with function
locally_linear_embedding or its object-oriented counterpart
LocallyLinearEmbedding, with the keyword method = 'modified'.
It requires n_neighbors > n_components.
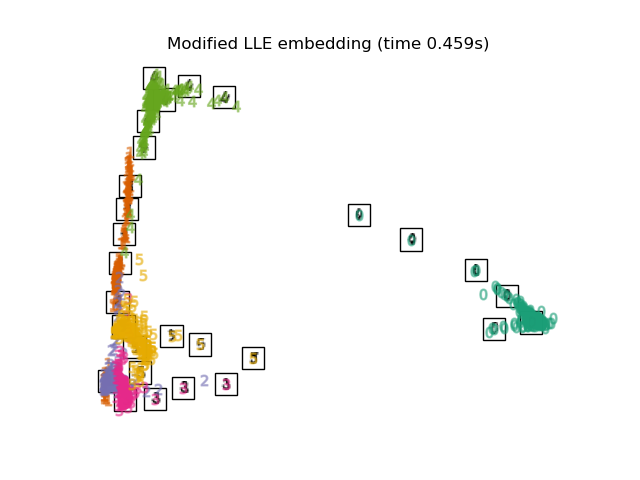
The MLLE algorithm comprises three stages: Nearest Neighbors Search. Same as standard LLE Weight Matrix Construction. Approximately
\(O[D N k^3] + O[N (k-D) k^2]\). The first term is exactly equivalent
to that of standard LLE. The second term has to do with constructing the
weight matrix from multiple weights. In practice, the added cost of
constructing the MLLE weight matrix is relatively small compared to the
cost of stages 1 and 3. Partial Eigenvalue Decomposition. Same as standard LLE The overall complexity of MLLE is
\(O[D \log(k) N \log(N)] + O[D N k^3] + O[N (k-D) k^2] + O[d N^2]\). \(N\) : number of training data points \(D\) : input dimension \(k\) : number of nearest neighbors \(d\) : output dimension
Complexity
Click for more details
2.2.5. Hessian Eigenmapping¶
Hessian Eigenmapping (also known as Hessian-based LLE: HLLE) is another method
of solving the regularization problem of LLE. It revolves around a
hessian-based quadratic form at each neighborhood which is used to recover
the locally linear structure. Though other implementations note its poor
scaling with data size, sklearn implements some algorithmic
improvements which make its cost comparable to that of other LLE variants
for small output dimension. HLLE can be performed with function
locally_linear_embedding or its object-oriented counterpart
LocallyLinearEmbedding, with the keyword method = 'hessian'.
It requires n_neighbors > n_components * (n_components + 3) / 2.
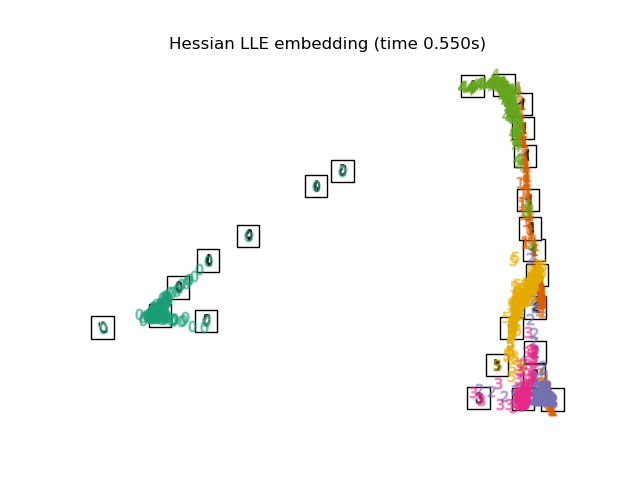
The HLLE algorithm comprises three stages: Nearest Neighbors Search. Same as standard LLE Weight Matrix Construction. Approximately
\(O[D N k^3] + O[N d^6]\). The first term reflects a similar
cost to that of standard LLE. The second term comes from a QR
decomposition of the local hessian estimator. Partial Eigenvalue Decomposition. Same as standard LLE The overall complexity of standard HLLE is
\(O[D \log(k) N \log(N)] + O[D N k^3] + O[N d^6] + O[d N^2]\). \(N\) : number of training data points \(D\) : input dimension \(k\) : number of nearest neighbors \(d\) : output dimension
Complexity
Click for more details
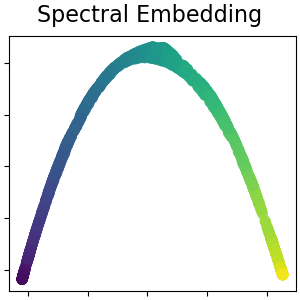
2.2.6. Spectral Embedding¶
Spectral Embedding is an approach to calculating a non-linear embedding.
Scikit-learn implements Laplacian Eigenmaps, which finds a low dimensional
representation of the data using a spectral decomposition of the graph
Laplacian. The graph generated can be considered as a discrete approximation of
the low dimensional manifold in the high dimensional space. Minimization of a
cost function based on the graph ensures that points close to each other on
the manifold are mapped close to each other in the low dimensional space,
preserving local distances. Spectral embedding can be performed with the
function spectral_embedding or its object-oriented counterpart
SpectralEmbedding.
The Spectral Embedding (Laplacian Eigenmaps) algorithm comprises three stages: Weighted Graph Construction. Transform the raw input data into
graph representation using affinity (adjacency) matrix representation. Graph Laplacian Construction. unnormalized Graph Laplacian
is constructed as \(L = D - A\) for and normalized one as
\(L = D^{-\frac{1}{2}} (D - A) D^{-\frac{1}{2}}\). Partial Eigenvalue Decomposition. Eigenvalue decomposition is
done on graph Laplacian The overall complexity of spectral embedding is
\(O[D \log(k) N \log(N)] + O[D N k^3] + O[d N^2]\). \(N\) : number of training data points \(D\) : input dimension \(k\) : number of nearest neighbors \(d\) : output dimension
Complexity
Click for more details
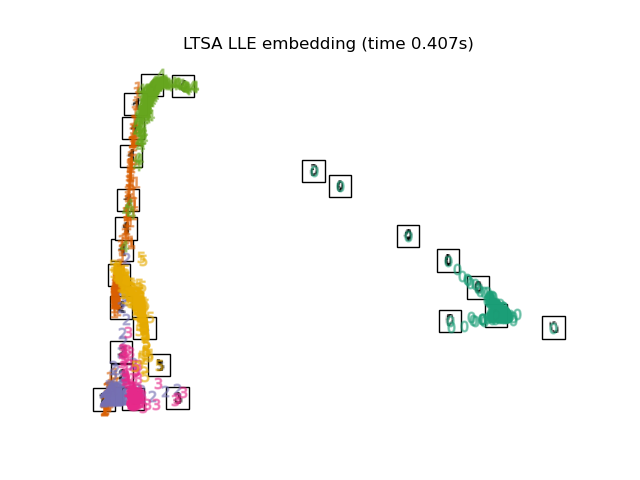
2.2.7. Local Tangent Space Alignment¶
Though not technically a variant of LLE, Local tangent space alignment (LTSA)
is algorithmically similar enough to LLE that it can be put in this category.
Rather than focusing on preserving neighborhood distances as in LLE, LTSA
seeks to characterize the local geometry at each neighborhood via its
tangent space, and performs a global optimization to align these local
tangent spaces to learn the embedding. LTSA can be performed with function
locally_linear_embedding or its object-oriented counterpart
LocallyLinearEmbedding, with the keyword method = 'ltsa'.
The LTSA algorithm comprises three stages: Nearest Neighbors Search. Same as standard LLE Weight Matrix Construction. Approximately
\(O[D N k^3] + O[k^2 d]\). The first term reflects a similar
cost to that of standard LLE. Partial Eigenvalue Decomposition. Same as standard LLE The overall complexity of standard LTSA is
\(O[D \log(k) N \log(N)] + O[D N k^3] + O[k^2 d] + O[d N^2]\). \(N\) : number of training data points \(D\) : input dimension \(k\) : number of nearest neighbors \(d\) : output dimension
Complexity
Click for more details
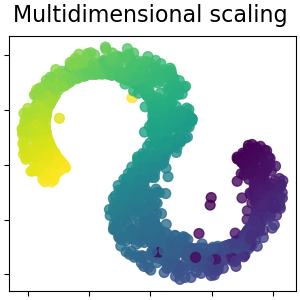
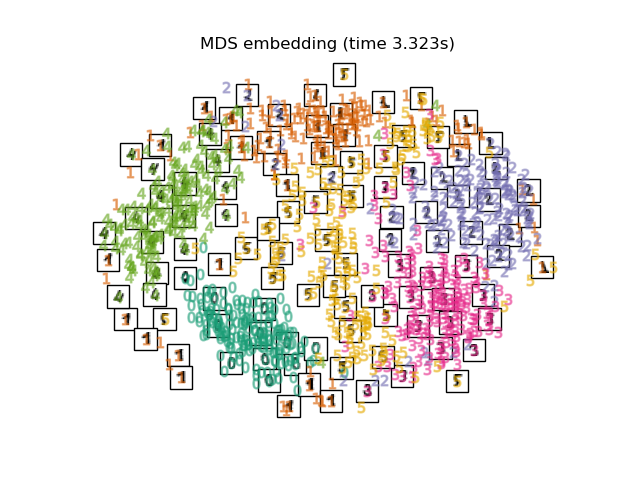
2.2.8. Multi-dimensional Scaling (MDS)¶
Multidimensional scaling
(MDS) seeks a low-dimensional
representation of the data in which the distances respect well the
distances in the original high-dimensional space.
In general, MDS is a technique used for analyzing similarity or
dissimilarity data. It attempts to model similarity or dissimilarity data as
distances in a geometric spaces. The data can be ratings of similarity between
objects, interaction frequencies of molecules, or trade indices between
countries.
There exists two types of MDS algorithm: metric and non metric. In
scikit-learn, the class MDS implements both. In Metric MDS, the input
similarity matrix arises from a metric (and thus respects the triangular
inequality), the distances between output two points are then set to be as
close as possible to the similarity or dissimilarity data. In the non-metric
version, the algorithms will try to preserve the order of the distances, and
hence seek for a monotonic relationship between the distances in the embedded
space and the similarities/dissimilarities.
Let \(S\) be the similarity matrix, and \(X\) the coordinates of the \(n\) input points. Disparities \(\hat{d}_{ij}\) are transformation of the similarities chosen in some optimal ways. The objective, called the stress, is then defined by \(\sum_{i < j} d_{ij}(X) - \hat{d}_{ij}(X)\)
The simplest metric Most commonly, disparities are set to \(\hat{d}_{ij} = b S_{ij}\).
Metric MDS
Click for more details
MDS model, called absolute MDS, disparities are defined by
\(\hat{d}_{ij} = S_{ij}\). With absolute MDS, the value \(S_{ij}\)
should then correspond exactly to the distance between point \(i\) and
\(j\) in the embedding point.
Non metric A trivial solution to this problem is to set all the points on the origin. In
order to avoid that, the disparities \(\hat{d}_{ij}\) are normalized. Note
that since we only care about relative ordering, our objective should be
invariant to simple translation and scaling, however the stress used in metric
MDS is sensitive to scaling. To address this, non-metric MDS may use a
normalized stress, known as Stress-1 defined as The use of normalized Stress-1 can be enabled by setting
Nonmetric MDS
Click for more details
MDS focuses on the ordination of the data. If
\(S_{ij} > S_{jk}\), then the embedding should enforce \(d_{ij} <
d_{jk}\). For this reason, we discuss it in terms of dissimilarities
(\(\delta_{ij}\)) instead of similarities (\(S_{ij}\)). Note that
dissimilarities can easily be obtained from similarities through a simple
transform, e.g. \(\delta_{ij}=c_1-c_2 S_{ij}\) for some real constants
\(c_1, c_2\). A simple algorithm to enforce proper ordination is to use a
monotonic regression of \(d_{ij}\) on \(\delta_{ij}\), yielding
disparities \(\hat{d}_{ij}\) in the same order as \(\delta_{ij}\).normalized_stress=True,
however it is only compatible with the non-metric MDS problem and will be ignored
in the metric case.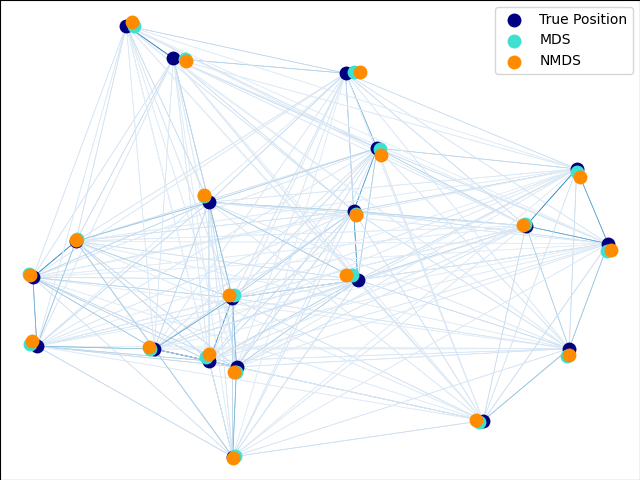
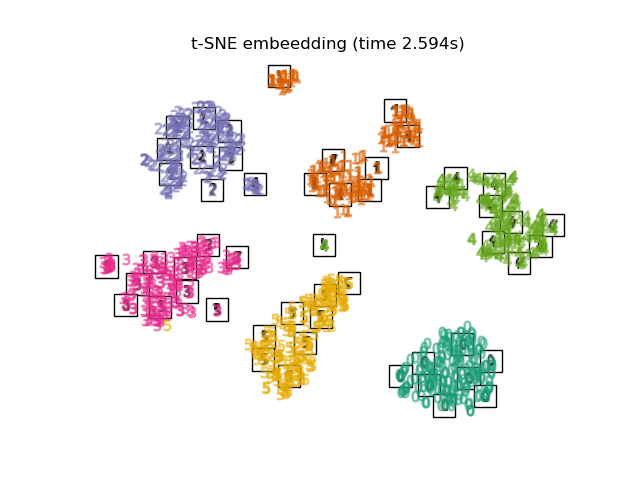
2.2.9. t-distributed Stochastic Neighbor Embedding (t-SNE)¶
t-SNE (TSNE) converts affinities of data points to probabilities.
The affinities in the original space are represented by Gaussian joint
probabilities and the affinities in the embedded space are represented by
Student’s t-distributions. This allows t-SNE to be particularly sensitive
to local structure and has a few other advantages over existing techniques:
Revealing the structure at many scales on a single map
Revealing data that lie in multiple, different, manifolds or clusters
Reducing the tendency to crowd points together at the center
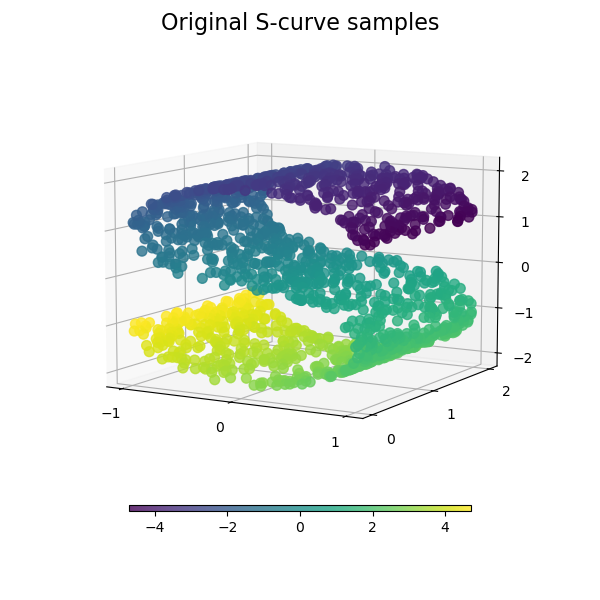
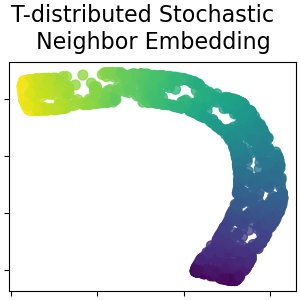
While Isomap, LLE and variants are best suited to unfold a single continuous low dimensional manifold, t-SNE will focus on the local structure of the data and will tend to extract clustered local groups of samples as highlighted on the S-curve example. This ability to group samples based on the local structure might be beneficial to visually disentangle a dataset that comprises several manifolds at once as is the case in the digits dataset.
The Kullback-Leibler (KL) divergence of the joint probabilities in the original space and the embedded space will be minimized by gradient descent. Note that the KL divergence is not convex, i.e. multiple restarts with different initializations will end up in local minima of the KL divergence. Hence, it is sometimes useful to try different seeds and select the embedding with the lowest KL divergence.
The disadvantages to using t-SNE are roughly:
t-SNE is computationally expensive, and can take several hours on million-sample datasets where PCA will finish in seconds or minutes
The Barnes-Hut t-SNE method is limited to two or three dimensional embeddings.
The algorithm is stochastic and multiple restarts with different seeds can yield different embeddings. However, it is perfectly legitimate to pick the embedding with the least error.
Global structure is not explicitly preserved. This problem is mitigated by initializing points with PCA (using
init='pca').
The main purpose of t-SNE is visualization of high-dimensional data. Hence,
it works best when the data will be embedded on two or three dimensions. Optimizing the KL divergence can be a little bit tricky sometimes. There are
five parameters that control the optimization of t-SNE and therefore possibly
the quality of the resulting embedding: perplexity early exaggeration factor learning rate maximum number of iterations angle (not used in the exact method) The perplexity is defined as \(k=2^{(S)}\) where \(S\) is the Shannon
entropy of the conditional probability distribution. The perplexity of a
\(k\)-sided die is \(k\), so that \(k\) is effectively the number of
nearest neighbors t-SNE considers when generating the conditional probabilities.
Larger perplexities lead to more nearest neighbors and less sensitive to small
structure. Conversely a lower perplexity considers a smaller number of
neighbors, and thus ignores more global information in favour of the
local neighborhood. As dataset sizes get larger more points will be
required to get a reasonable sample of the local neighborhood, and hence
larger perplexities may be required. Similarly noisier datasets will require
larger perplexity values to encompass enough local neighbors to see beyond
the background noise. The maximum number of iterations is usually high enough and does not need
any tuning. The optimization consists of two phases: the early exaggeration
phase and the final optimization. During early exaggeration the joint
probabilities in the original space will be artificially increased by
multiplication with a given factor. Larger factors result in larger gaps
between natural clusters in the data. If the factor is too high, the KL
divergence could increase during this phase. Usually it does not have to be
tuned. A critical parameter is the learning rate. If it is too low gradient
descent will get stuck in a bad local minimum. If it is too high the KL
divergence will increase during optimization. A heuristic suggested in
Belkina et al. (2019) is to set the learning rate to the sample size
divided by the early exaggeration factor. We implement this heuristic
as “How to Use t-SNE Effectively”
provides a good discussion of the effects of the various parameters, as well
as interactive plots to explore the effects of different parameters.
Optimizing t-SNE
Click for more details
learning_rate='auto' argument. More tips can be found in
Laurens van der Maaten’s FAQ (see references). The last parameter, angle,
is a tradeoff between performance and accuracy. Larger angles imply that we
can approximate larger regions by a single point, leading to better speed
but less accurate results.
The Barnes-Hut t-SNE that has been implemented here is usually much slower than
other manifold learning algorithms. The optimization is quite difficult
and the computation of the gradient is \(O[d N log(N)]\), where \(d\)
is the number of output dimensions and \(N\) is the number of samples. The
Barnes-Hut method improves on the exact method where t-SNE complexity is
\(O[d N^2]\), but has several other notable differences: The Barnes-Hut implementation only works when the target dimensionality is 3
or less. The 2D case is typical when building visualizations. Barnes-Hut only works with dense input data. Sparse data matrices can only be
embedded with the exact method or can be approximated by a dense low rank
projection for instance using Barnes-Hut is an approximation of the exact method. The approximation is
parameterized with the angle parameter, therefore the angle parameter is
unused when method=”exact” Barnes-Hut is significantly more scalable. Barnes-Hut can be used to embed
hundred of thousands of data points while the exact method can handle
thousands of samples before becoming computationally intractable For visualization purpose (which is the main use case of t-SNE), using the
Barnes-Hut method is strongly recommended. The exact t-SNE method is useful
for checking the theoretically properties of the embedding possibly in higher
dimensional space but limit to small datasets due to computational constraints. Also note that the digits labels roughly match the natural grouping found by
t-SNE while the linear 2D projection of the PCA model yields a representation
where label regions largely overlap. This is a strong clue that this data can
be well separated by non linear methods that focus on the local structure (e.g.
an SVM with a Gaussian RBF kernel). However, failing to visualize well
separated homogeneously labeled groups with t-SNE in 2D does not necessarily
imply that the data cannot be correctly classified by a supervised model. It
might be the case that 2 dimensions are not high enough to accurately represent
the internal structure of the data.
Barnes-Hut t-SNE
Click for more details
TruncatedSVD
2.2.10. Tips on practical use¶
Make sure the same scale is used over all features. Because manifold learning methods are based on a nearest-neighbor search, the algorithm may perform poorly otherwise. See StandardScaler for convenient ways of scaling heterogeneous data.
The reconstruction error computed by each routine can be used to choose the optimal output dimension. For a \(d\)-dimensional manifold embedded in a \(D\)-dimensional parameter space, the reconstruction error will decrease as
n_componentsis increased untiln_components == d.Note that noisy data can “short-circuit” the manifold, in essence acting as a bridge between parts of the manifold that would otherwise be well-separated. Manifold learning on noisy and/or incomplete data is an active area of research.
Certain input configurations can lead to singular weight matrices, for example when more than two points in the dataset are identical, or when the data is split into disjointed groups. In this case,
solver='arpack'will fail to find the null space. The easiest way to address this is to usesolver='dense'which will work on a singular matrix, though it may be very slow depending on the number of input points. Alternatively, one can attempt to understand the source of the singularity: if it is due to disjoint sets, increasingn_neighborsmay help. If it is due to identical points in the dataset, removing these points may help.
See also
Totally Random Trees Embedding can also be useful to derive non-linear representations of feature space, also it does not perform dimensionality reduction.